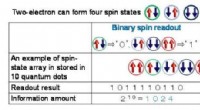
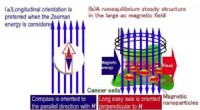
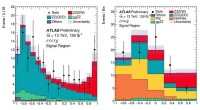
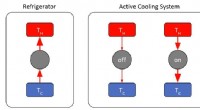
The new technique, called "self-consistent-field density functional theory with dynamical screening" (SCF-DFT+DS), reduces the computational cost of calculating the electron density in a material by up to 90% compared to conventional methods. This makes it possible to perform calculations on much larger systems, such as those found in real-world materials.
"SCF-DFT+DS is a significant breakthrough in the field of materials science," said Argonne scientist Giulia Galli, who led the research team. "It will enable us to study a wider range of materials and phenomena, and design new materials with improved properties for a variety of applications."
The SCF-DFT+DS technique is based on a reformulation of the density functional theory (DFT) equations. DFT is a widely used method for calculating the electronic structure of materials, but it can be computationally expensive for large systems. The new technique uses a simplified representation of the electron-electron interaction, which reduces the computational cost without sacrificing accuracy.
The research team tested the new technique on several systems, including semiconductors, metals, and insulators. They found that SCF-DFT+DS produced results that were in excellent agreement with conventional DFT, but at a fraction of the computational cost.
"SCF-DFT+DS is a powerful new tool that will open up new possibilities for materials research," said Galli. "We are excited to explore its potential and use it to design new materials for a cleaner, more sustainable future."
The research was published in the journal Physical Review Letters.