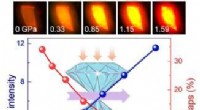
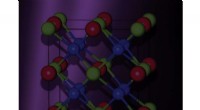
The research team employed density functional theory (DFT) calculations to investigate the behavior of water molecules in the presence of ions. DFT is a powerful tool for simulating the electronic structure of materials and molecules. The team focused on two specific systems: the solvation of lithium ions (Li+) and the interaction of water molecules with a lithium-oxygen (Li-O) surface.
In the case of Li+ solvation, the DFT calculations revealed that water molecules form a highly ordered hydration shell around the lithium ion. This ordered structure results from the strong electrostatic interactions between the positively charged lithium ion and the negatively charged oxygen atoms of water molecules. The hydration shell effectively shields the lithium ion from interacting with other ions or molecules in the solution, which is crucial for stabilizing the ion and facilitating its transport in electrochemical reactions.
Furthermore, the researchers investigated the interaction of water molecules with a Li-O surface, which represents the electrode surface in a lithium-ion battery. The DFT calculations showed that water molecules form strong hydrogen bonds with the oxygen atoms on the surface, creating a water network that effectively blocks the migration of lithium ions from the electrode. This blocking effect is responsible for the passivation of the electrode surface and the reduction of battery performance over time.
Overall, the theoretical framework developed by the research team provides a comprehensive understanding of the role of water molecules in material synthesis and manufacturing processes involving ions. The findings contribute to the rational design and optimization of electrochemical reactions and battery systems, enabling more efficient and durable materials and devices.