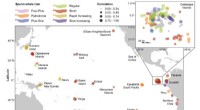
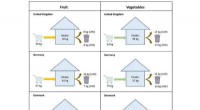
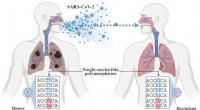
- Input: Single-cell RNA-seq data (count matrix)
- Quality Control (QC): Remove low-quality cells and genes
- Data Normalization: Normalize the data to correct for technical biases
2. Clustering
- Perform clustering on the normalized data to identify cell clusters
- Different clustering methods can be used (e.g., k-means, hierarchical clustering, Louvain)
3. Marker Gene Identification
- For each cluster:
- Calculate the mean expression of each gene across cells in the cluster
- Compare the mean expression of genes in the cluster to that in other clusters
- Identify genes that are highly expressed in the cluster compared to other clusters
4. Marker Gene Validation
- Additional criteria can be applied to select marker genes:
- Fold change: Consider genes with a high fold change between the cluster and other clusters
- Statistical significance: Use statistical tests (e.g., t-test, Wilcoxon test) to assess the significance of expression differences
- Specificity: Ensure that marker genes are selectively expressed in the cluster of interest
5. Interpretation and Visualization
- Analyze the functions and pathways associated with the identified marker genes
- Generate heatmaps, volcano plots, or other visualizations to present the marker genes and their expression patterns
6. Validation in Independent Datasets (optional)
- To increase confidence, validate the identified marker genes in an independent dataset if available.