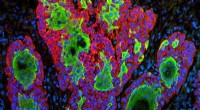
Lysosomes are membrane-bound organelles that contain hydrolytic enzymes that break down a variety of molecules, including proteins, lipids, and carbohydrates. Lysosomes are essential for maintaining cellular homeostasis, but they can also be dangerous if they rupture, releasing their contents into the cytoplasm. To prevent this, cells have a number of mechanisms in place to ensure that damaged lysosomes are quickly identified and removed.
One of the most important mechanisms for selecting damaged lysosomes for clearance is the process of ubiquitination. Ubiquitin is a small protein that can be attached to other proteins to mark them for degradation. When a lysosome is damaged, ubiquitin ligases are recruited to the site of damage and they attach ubiquitin molecules to the lysosomal membrane. This marks the lysosome for recognition by autophagy receptors, which are proteins that bind to ubiquitin and target the lysosome for delivery to the autophagosome, a double-membrane vesicle that fuses with the lysosome to deliver its contents for degradation.
In a recent study, researchers at the University of California, San Francisco used molecular tags to track the fate of damaged lysosomes in cells. They found that ubiquitination is not the only mechanism that can mark damaged lysosomes for clearance. They also identified a number of other molecular tags that can be used to select damaged lysosomes, including:
* HMGB1: HMGB1 is a protein that is released from the nucleus when cells are stressed. HMGB1 can bind to the lysosomal membrane and recruit autophagy receptors.
* LC3: LC3 is a protein that is involved in autophagosome formation. LC3 can also bind to the lysosomal membrane and recruit autophagy receptors.
* p62: p62 is a protein that is involved in the aggregation of damaged proteins. p62 can bind to the lysosomal membrane and recruit autophagy receptors.
The researchers found that these molecular tags work together to ensure that damaged lysosomes are quickly identified and removed from cells. This process is essential for maintaining cellular homeostasis and preventing the development of lysosomal storage diseases.
Implications for lysosomal storage diseases
Lysosomal storage diseases are a group of genetic disorders that are caused by mutations in genes that encode proteins involved in lysosomal function. These mutations lead to the accumulation of undigested material in lysosomes, which can damage cells and tissues. Lysosomal storage diseases are often fatal, and there is currently no cure.
The research described above provides new insights into the mechanisms that cells use to select and remove damaged lysosomes. This knowledge could lead to the development of new therapies for lysosomal storage diseases. For example, it may be possible to develop drugs that inhibit the ubiquitination of lysosomes or that block the binding of autophagy receptors to lysosomes. These drugs could help to prevent the accumulation of undigested material in lysosomes and slow the progression of lysosomal storage diseases.